


Cando a comezos deste 2020 unha nova enfermidade respiratoria se comezaba a propagar pola provincia chinesa de Hubei, e a meirande parte da sociedade escoitaba aquilo como un pequeno e distante ruído de fondo, o xenetista Antonio Salas comezou a interesarse por aquel novo virus, bautizado como SARS-CoV-2. Este mércores, máis de catro meses despois, Salas, xunto a Federico Martinón e os seus compañeiros de investigación da USC e o IDIS de Santiago, poñían a disposición da comunidade científica un artigo preliminar, á espera de superar todo o proceso de revisión que require a súa publicación nunha revista, que narra a viaxe ao longo do tempo e o espazo da minúscula partícula que dobregou a medio planeta. “Para min este traballo ten un compoñente emocional moi grande, porque foron moitas horas, noites sen case durmir, para poder xuntar todas as pezas, e cando ves que cadran, e que por riba descobres algo como a relevancia dos supercontaxiadores, a satisfacción que sentes é enorme”, resume o científico galego.
Desde os comezos da súa carreira, Salas especializouse na xenética de poboacións, unha disciplina que mestura a historia e os movementos da humanidade desde hai miles de anos ata a actualidade, coas marcas que as migracións e os encontros entre distintos pobos van deixando no ADN. A partir de aí, por exemplo, é posible botar luz sobre a orixe dos escravos dos Estados Unidos, os indios taínos ou dos afrobolivianos. E desde hai uns meses, o investigador centrou os seus esforzos en coñecer a circulación do SARS-CoV-2. “Nós non somos expertos en virus, pero si podemos analizar o seu xenoma e os individuos que o transmiten”, resume.

“Coa xenómica podemos narrar o ‘conto’ do coronavirus”
Desde a perspectiva da xenética evolutiva, os investigadores galegos decidiron contar a historia da propagación. “Coa xenómica podemos narrar o ‘conto’ do coronavirus”, resume o autor principal da investigación. “Unha das cousas máis importantes para coñecer o ‘becho’ é coñecer a súa taxonomía. Linneo xa o sabía ben, e non fixo o que fixo por casualidade, clasificar resulta esencial para entender. Co virus podemos facer algo parecido: saber como se relacionan unhas liñaxes con outras, de onde veñen, cara a onde van, en que cantidades están presentes, etc”.
150 millóns de letras
Grazas ás técnicas de computación que permiten ler a gran velocidade o código xenético do virus, os investigadores foron detectando as pequenas variacións que o virus ía experimentando ao pasar de organismo en organismo. Traballaron con preto de 5.000 mostras distintas, o que supón ler uns 150 millóns de letras, coas súas pequenas ‘grallas’, as marcas que cada hóspede vai deixando no patóxeno antes de transmitilo a outro, e así sucesivamente. O mapa baixo estas liñas amosa como se distribuíron as dúas principais variantes do virus (A e B) e os seus ‘descendentes’ a través do mundo.

E foi así como nun traballo que xa era prometedor apareceu o ‘eureka‘: os supercontaxiadores. “Non os estabamos buscando, pero ao analizar a filoxeografía (as diferenzas e semellanzas de cada mostra e a súa distribución polo planeta) atopamos múltiples casos nos que unha cepa concreta, por algún motivo que aínda non podemos precisar -aínda que seguimos investigándoo-, se propagaba moito máis cás outras.
Por que hai supercontaxiadores?
Os expertos de diferentes ramas (epidemioloxía, matemáticas, inmunoloxía, viroloxía) coinciden no papel clave desas persoas que, a veces asintomáticas, foron capaces de espallar a gran escala o SARS-CoV-2. Pero os motivos, por agora, non están claros. “Poden ser moitísimos factores os que conflúan: algúns son puramente biolóxicos, pero seguro que tamén xogan un papel clave o comportamento das persoas e a súa mobilidade, os períodos de incubación máis ou menos longos ou a propia carga viral. Na xenética forense sábese desde hai moito tempo que, por exemplo, unhas persoas son mellores transmisoras de células epiteliais (da pel) ca outras, e desta maneira poden transmitir o seu ADN a terceiras persoas coas que non tivo contacto a través de ‘vectores’ directos”, engade Salas.
O estudo suxire, por exemplo, que se debería afondar na análise do xenoma dos supercontaxiadores, no caso de poder atopalos, para ver se teñen unha configuración determinada que aumente o risco de transmisión do virus a outras persoas e o propio comportamento do virus.
O inicio dos focos en Hubei
A análise que realizaron os investigadores galegos permite tamén establecer a data aproximada da primeira infección do patóxeno que contaxiou millóns de persoas no planeta, o chamado “devanceiro común máis recente”. Sitúano arredor do 12 de novembro de 2019, e “asumindo un período máximo de incubación en humanos de 24 días, o virus podería comezar a infectar ás primeiras persoas en Hubei de forma ‘silenciosa‘ ata o final de novembro de 2019, sendo detectado polas autoridades sanitarias chinesas a comezos ou mediados de decembro”, di o artigo. Para o 30 de decembro, o día antes de que as autoridades chinas alertaran de forma oficial dos brotes, é moi probable que algúns “supercontaxiadores” xa estiveran propagando, sen sabelo, a pandemia.
Desde ese momento, o ‘conto‘ que Salas e o seu equipo narran a través da xenética achega datos moi relevantes, que están espertando un gran interese internacional despois da publicación do artigo. Unha das datas clave é o 20 de xaneiro; a partir dese día, a ‘explosión’ de brotes é patente por un gran aumento nos cambios do xenoma do SARS-CoV-2. “A xenética é un reloxo case perfecto, porque a través do estudo desas mostras do virus podemos determinar a data na que se infectou moita xente en Asia”, explica. Entón, compararon a ‘explosión‘ na variabilidade do virus co número de casos, e atoparon máis cousas, que se manifestan no gráfico baixo estas liñas (a orde cronolóxica é de dereita a esquerda).

Mentres que a liña azul amosa a variabilidade no xenoma, a curva laranxa amosa o número de casos detectados. O desprazamento da curva laranxa cara á esquerda podería indicar, segundo Salas, dúas hipóteses: que o período de incubación fose máis longo, ou que houbera bastantes máis casos dos que realmente se documentaron.
Ademais, o mapa xenético do SARS-CoV-2 vese alterado por outro factor, o do confinamento: “Cando as autoridades limitan os movementos da poboación en Asia, a finais de xaneiro, o virus deixa de mutar, ata que a metade de febreiro aparecen os primeiros brotes fóra de Asia e volve aumentar a variabilidade. É de agardar que aquí teña pasado o mesmo”.
B3a: A liñaxe ibérica
Dentro das distintas cepas que chegaron desde Asia a Europa a comezos de ano, hai unha que distingue á península ibérica do resto, a B3a. No resto de Europa, a liñaxe A2a foi a máis numerosa, estendéndose despois a África e América, pero en España destaca a presenza da outra liñaxe. E parece que chegou, segundo explica Salas, a través dun supercontaxiador que puido ter un papel clave nos máis de 230.000 casos detectados no país e as 28.000 mortes.
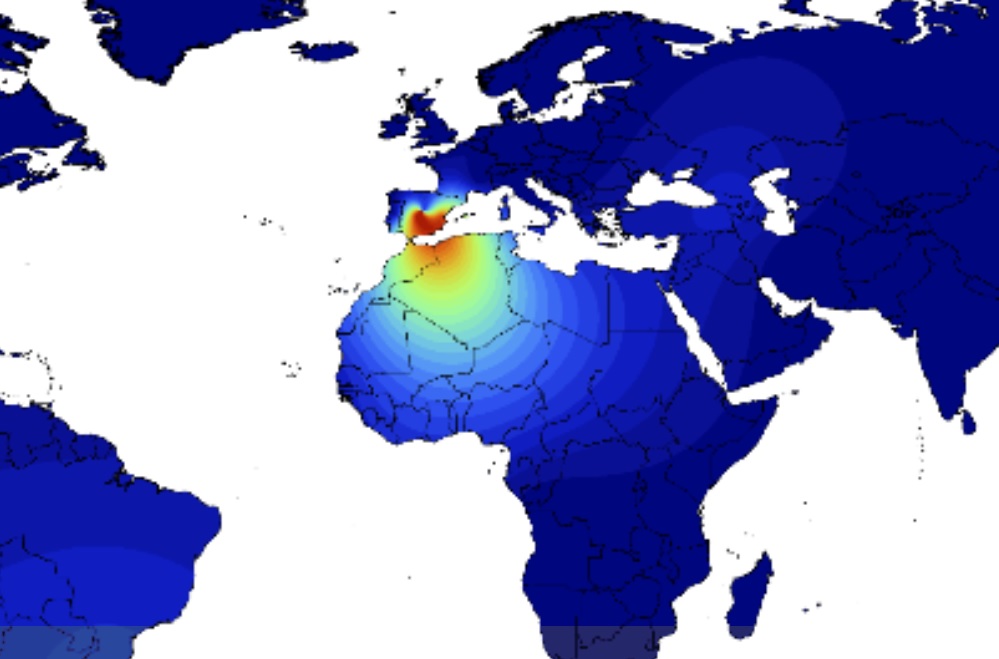
Os mapas realizados para cada unha das liñaxes amosan o especial impacto da B3a en España e o norte de África. E posteriormente, tamén se viu que os ‘descendentes‘ desa cepa viaxaron a América do Sur. Por que esta, e non outra? Por agora, non se pode explicar. “Existen certos fenómenos na xenética evolutiva que teñen que ver cos movementos dos individuos e cos propios virus que os acompañan; ás veces son aleatorios (tecnicamente reciben o nome de deriva xénica), e outras veces son fenómenos selectivos (selección natural positiva, purificadora, etc.), pero neste caso, os datos apuntan a que foi o azar o que acabou marcando o camiño: unha persoa infectada que viaxa a Europa desde Asia cunha determinada liñaxe e que, por unha sucesión de casualidades, acaba sendo o vector de contaxio para moitísima xente”.
Incógnitas
Coa publicación do artigo no repositorio Biorxiv, os investigadores galegos pretenden compartir os avances coa comunidade científica para enriquecer o seu traballo cos comentarios e valoracións dos colegas. Ao tempo, o equipo segue mergullándose na historia do SARS-CoV-2, e Salas avanza que probablemente nas vindeiras semanas obterán novos datos relevantes sobre o xenoma do coronavirus.
Con todo, e a pesar das especulacións sobre a maior ou menor gravidade de determinadas cepas, a análise xenética non parece consolidar esta posibilidade. “Por agora non temos evidencia de que haxa cepas máis agresivas ca outras”. Do mesmo xeito, as variacións que están atopando, engade, poderían axudar á hora de perfeccionar a posible vacina e adiantarse ás dificultades que poidan xurdir: “Ao mirar o xenoma, e buscar as mutacións que se van acumulando, observamos que había moi poucos ocos ‘libres’ de alteracións, o que podería traducirse nunha maior dificultade para atopar unha vacina duradeira, e igual teriamos que inmunizarnos cunha ‘fórmula’ distinta cada certo tempo, como coa gripe”, conclúe Antonio Salas.
Supercontaxiadores da Covid-19: esta é a clave que revelan científicos galegos









